正常上班时间门急诊咨询:0731-83929900(天心阁)/84762791(马王堆)/81866120(岳麓山)
非正常上班时间门急诊咨询:0731-82278048(天心阁)/84731731(马王堆)/19892801600(岳麓山)
非正常上班时间门急诊咨询:0731-82278048(天心阁)/84731731(马王堆)/19892801600(岳麓山)






机构办安全性事件接收邮箱:[email protected]
系统上报流程(下图):CTMS(//ethics.tonoinfo.com/)-项目中心-安全模块(SAE/SUSAR/DSUR)-新增(首次报告/随访报告/总结报告)
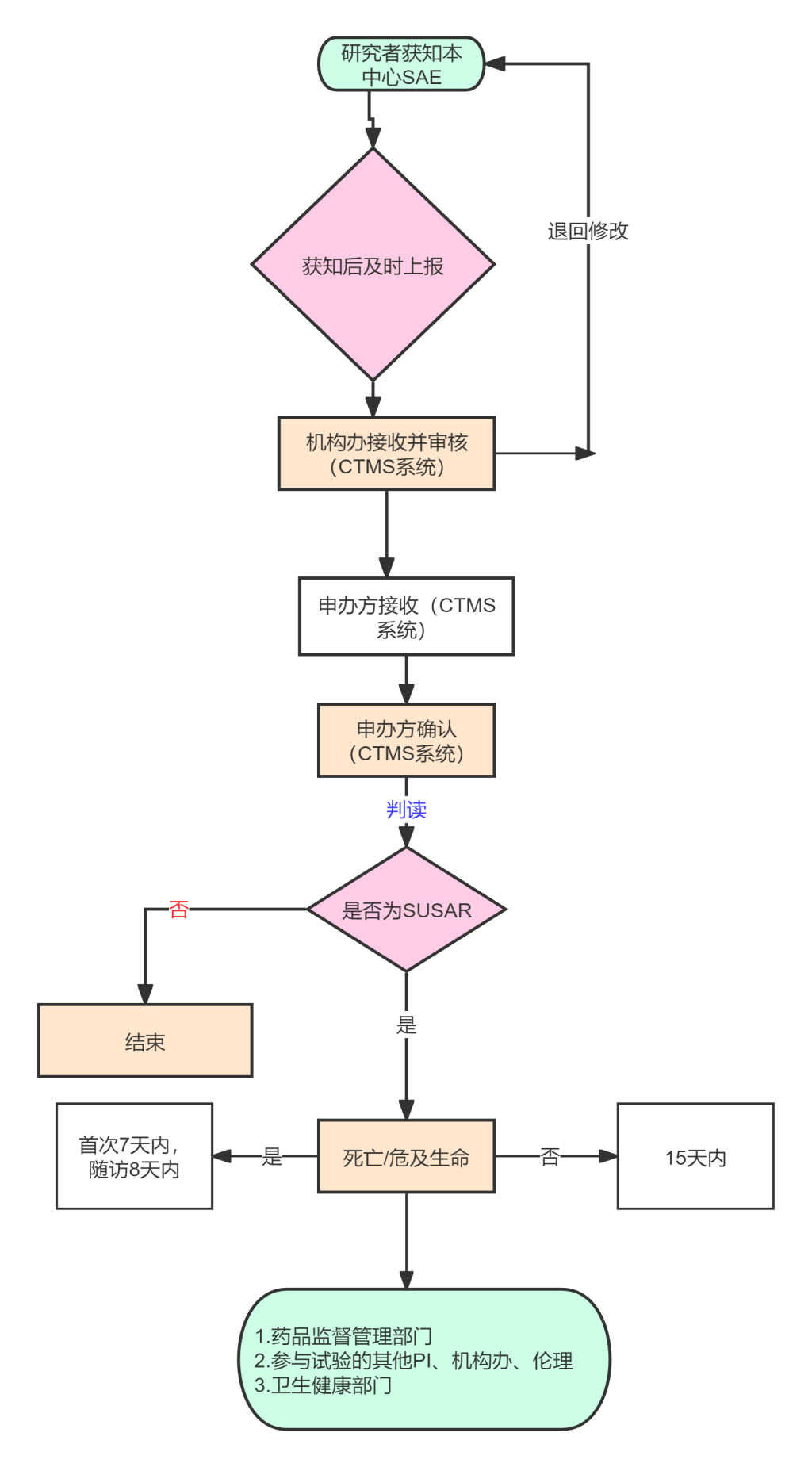
(一)严重不良事件(SAE)的上报
本中心SAE上报:除试验方案或者其他文件(如研究者手册)中规定不需立即报告的严重不良事件外,研究者应当在首次获知SAE后立即完成以下报告:
研究者在系统中填报SAE,从系统发送至捷克论坛 机构、伦理及申办方,星号项必填,附件上传经研究者签字确认的SAE报告PDF版。随后及时补充“随访报告”或“总结报告”,并按上述流程报告至各相关方。
非本中心SAE上报:根据方案要求进行报告。
(二)本中心可疑且非预期的严重不良反应(SUSAR)
经申办者评估为可疑且非预期的严重不良反应(SUSAR),完成以下报告:
1. 申办者递交研究者,经研究者审核签字;
2. 研究者在系统中填报SUSAR,同时发送至机构办、伦理;
报告时限应当遵循《药物临床试验期间安全性数据快速报告标准和程序》要求:
1)对于致死或危及生命的SUSAR,应在首次获知后尽快报告,最迟不得超过7天,并在随后的8天内报告、完善随访信息。(注:申办者首次获知当天为第0天。);
2)对于非致死或危及生命的SUSAR,应在首次获知后尽快报告,最迟不得超过15天;
3)申办者在首次报告后,应继续跟踪严重不良反应,以随访报告的形式及时报送有关新信息或对前次报告的更改信息等,报告时限为获得新信息起15天内。
(三)非本中心SUSAR
申办者收到外院安全性信息后,应通过系统每3个月进行1次填报。
(四)研发期间安全性更新报告(DSUR)的上报
申办者定期汇总DSUR并向捷克论坛 报告,原则上报告周期不超过一年。
文件要求(至少应包括):①严重不良反应(SAR)累计汇总表;②报告周期内境内死亡受试者列表;③报告周期内境内因任何不良事件而退出临床试验的受试者列表;④报告周期内发生的药物临床试验方案变更或者临床方面的新发现、非临床或者药学的变化或者新发现总结表;⑤下一报告周期内总体研究计划概要。