正常上班时间门急诊咨询:0731-83929900(天心阁)/84762791(马王堆)/81866120(岳麓山)
非正常上班时间门急诊咨询:0731-82278048(天心阁)/84731731(马王堆)/19892801600(岳麓山)
非正常上班时间门急诊咨询:0731-82278048(天心阁)/84731731(马王堆)/19892801600(岳麓山)






系统上报流程(下图):CTMS(//ethics.tonoinfo.com/)-
项目中心-安全模块(SAE/SUSAR/DSUR)-新增(首次报告/随访报告/总结报告)
报告模板(点击):医疗器械/体外诊断试剂临床试验严重不良事件报告表
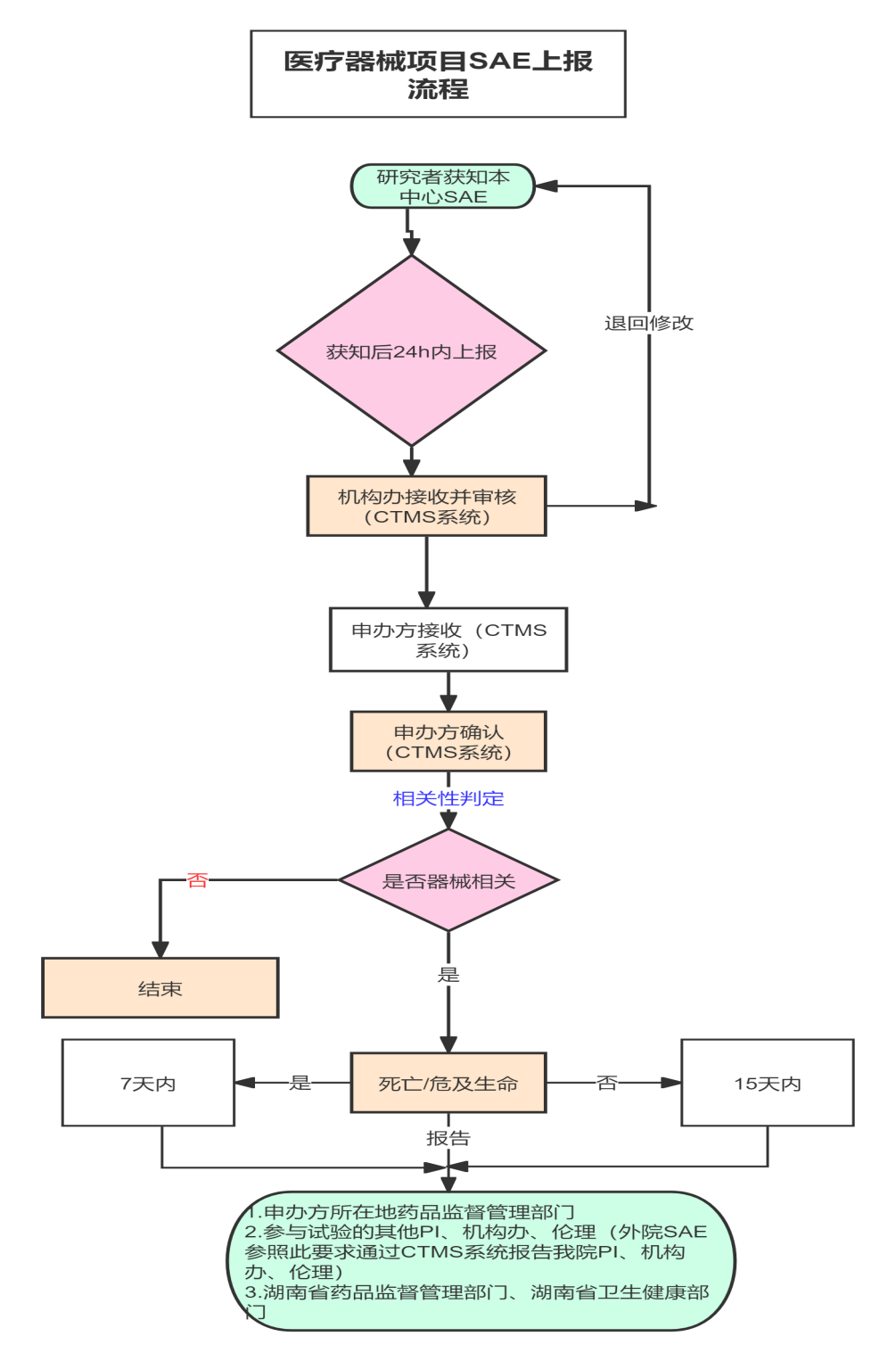
(一)报告流程及要求
本中心发生的SAE
1.研究者在获知SAE后,在24小时内通过CTMS系统填写并报告至申办者、机构办公室、伦理,星号项必填,附件上传医疗器械临床试验SAE报告表(研究者签字确认PDF版),报告表格均采用《医疗器械临床试验质量管理规范》(2022年)中的范本,随后及时补充“随访报告”或“总结报告”,并按上述流程报告至各相关方。
2.申办者/CRO在对死亡或者危及生命的临床试验医疗器械相关严重不良事件7日内、对非死亡或者非危及生命的试验医疗器械相关严重不良事件和其他严重安全性风险信息15日内向其他医疗器械临床试验机构、主要研究者、伦理委员会报告,同时向申办者所在地省、自治区、直辖市药品监督管理部门报告,向湖南省药品监督管理部门和卫生健康管理部门报告。
非本院发生SAE
申办者应当在获知死亡或者危及生命的临床试验医疗器械相关严重不良事件后7日内、获知非死亡或者非危及生命的试验医疗器械相关严重不良事件和其他严重安全性风险信息后15日内,向本临床试验机构和伦理委员会办公室报告。
(二)报告内容及要求:
1.详细处理情况除包含常规内容外,必须明确以下内容:a)描述受试者参加医疗器械临床试验情况;b)描述试验医疗器械使用情况,对有源和无源医疗器械应当描述器械具体操作使用情况,出现的非预期结果,(可能)对受试者造成的伤害,采取的救治措施以及结果等。对体外诊断医疗器械,应当描述受试者诊疗信息(如疾病情况、用药情况等)、样本检测过程与结果、发现的异常情况、采取的措施、最终结果判定、对临床诊疗的影响等;c)描述严重不良事件发生与处理情况;d)事件与关系的判定依据。
2.研究者应按照受试者发生的不良事件的具体情况和既往病史、伴随疾病情况以及伴随用药等情况进行综合分析,判断事件与医疗器械的关系。判断原则为:
a)与试验医疗器械有关:(1)两者存在合理时间关系;(2)试验器械已知风险或者可以用试验器械的机理去解释;(3)停止使用后伤害减轻或消失;(4)再次使用后伤害再次出现;(5)无法用其他影响因素解释。同时满足其中五条判断为“肯定有关”,满足其中两条判断为“可能有关”;
b)与试验医疗器械无关:(1)两者不存在合理时间关系;(2)该不良事件为该试验医疗器械不可能导致的事件类型;(3)该不良事件可用合并用械/药、受试者病情进展、其他治疗影响来解释。同时满足三条判断为“肯定无关”,满足其中一条判断为“可能无关”。
3.当出现研究方案中规定SAE与研究终点的确定需要第三方裁定的情况,应在按照研究方案中规定的流程,进行相关裁定的同时进行SAE的报告;如裁定结果不为SAE,研究者应在获知裁定结果的24小时内参照相关要求进行SAE的撤销或者修正报告。